
1、歐盟RoHS2.0(2011/65/EU)于2011年7月1日正式在歐盟官方公報上發布,醫療設備和監控設備及其零部件自2014年7月22日起應符合ROHS2.0,體外診斷醫療設備及其零部件自2016年7月22日起應符合ROHS2.0。
2、出口歐盟的企業要注意了,如果其產品落在RoHS的范圍,沒有符合RoHS2.0的認證,等同于不滿足歐盟法規的認證。簡單說,不符合RoHS指令的有源醫療器械產品,不能加貼CE標志。
3、企業新研發或目標市場在歐盟的企業,研發過程就應注意RoHS要求的實現。任何重大法規或標準實施前,都給行業或企業留有過渡期。建議企業珍惜這個過渡期,未雨綢繆。
-----------------------------------
RoHS指令對醫療器械的環保要求及應對措施
一、醫療器械適用的RoHS2.0要求
1. 需要符合RoHS要求的醫療器械
我們常說的RoHS即歐盟《限制某些有害物質在電子電器設備中的使用指令》,現在該指令已是第2版,所以業界一般將其簡稱為RoHS2.0。
RoHS適用的產品對象是所有進入歐盟市場的“電子電器設備”。在新版的RoHS2.0中,醫療器械屬于新納入的產品類別,需要符合RoHS2.0要求。
需要明確的是,并非所有醫療器械都需要滿足RoHS要求,只有那些屬于RoHS2.0指令定義的“電子電器設備”范圍的醫療器械,才需要滿足RoHS要求。
RoHS2.0對“電子電器設備”的定義為:指依賴電流或電磁場才能正常工作的設備,以及用于產生、轉換和測量上述電流或電磁場的設備,且設計使用電壓交流不超過1000伏、直流不超過1500伏。
所以,只要符合上述定義的醫療器械,都屬于需要符合RoHS要求的“電子電器設備”。請注意,RoHS2.0將“植入式醫療器械”排除在外,即使它們符合上述“電子電器設備”的定義,這類醫療器械也無需滿足RoHS2.0的要求。
通常,業界將耗電的醫療器械簡稱為“有源醫療器械”。雖然這個不很準確,為行文方便,本文還是采用這個術語。
2. RoHS2.0對有源醫療器械的生效時間
RoHS指令對不同類別的電子電器設備規定了不同的生效時間。
對于醫療器械的生效時間,RoHS2.0進一步分別針對一般的醫療器械(符合歐盟93/42/ EEC 指令第1(2)款第a點的定義的)和“體外診斷醫療器械”(符合歐盟98/79/EC指令第1(2)款第b點的定義的)進行了規定。
對于一般醫療器械,RoHS指令生效的時間是2014年7月22日,即從2014年7月22日開始投放歐盟市場的一般醫療器械必須符合RoHS2.0的要求了。
對于“體外診斷醫療器械”,其生效時間則為2016年7月22日。
所以,現在我們必須關注的是普通有源醫療器械的RoHS符合性。
3. RoHS2.0對有源醫療器械的適用要求
RoHS2.0對有源醫療器械的適用要求羅列如下: 產品中含有的“鉛”、“鎘”、“汞”、“六價鉻”、“多溴聯苯”、“多溴聯苯醚”等六類物質不能超過限值。對這六類物質的限值除了“鎘”為“0.01%”外,其余五類的都是“0.1%”。而且,對這些有害物質的限值都是從“均質材料”層次進行的,極為嚴格。這個要求是RoHS2.0指令的基本要求。
此外,RoHS2.0對制造商還有如下要求:
制造商要按照第768/2008/EC決定的附件II的模式A制訂技術文檔,實施內部生產控制程序,或確保其得到實施;
制造商應制訂歐盟合格聲明,并在成品上加貼CE標志;
制造商應留存技術文檔和歐盟合格聲明到電子電氣設備投放市場后10年;
為保持符合性,制造商應確保系列生產執行該程序。應充分考慮產品設計變更、特性變更,以及對電子電氣設備合格聲明引用過的協調標準或技術規范變更;
制造商應登記不符合的電子電氣設備和產品召回,并通知相關經銷商;
制造商應確保自己的電子電氣設備帶有型號、批次和序列號或其他有助于識別產品的要素;在產品的形狀或性質不允許時,在包裝或產品隨附文件中提供;
制造商應將公司名稱、注冊的商標名稱或注冊商標標識及聯系地址在電子電氣設備上標明,不可行時,在產品包裝或隨附文件中提供。地址一定要指明能夠聯系到制造商的一個點;在歐盟其他適用法規中包含至少同等嚴格程度的加貼制造商名稱和地址的條款時,適用那些條款;
當制造商認為或有理由相信投放市場的產品不符合本指令時,應立即采取必要的糾正措施使該電子電氣設備符合、適宜時撤回或召回;而且,應立即將相關情況通知產品所投放成員國的主管部門,特別應告知不符合情況、應采取糾正措施的詳細信息;
應主管部門的合理要求,制造商應以主管部門能理解的語言向其提供可用于證明電子電氣設備符合本指令的所有信息和文件。應主管部門要求,他們應與主管部門合作,針對其投放市場的產品采取確保符合本指令條款的任何措施。
二、醫療器械企業CE認證與RoHS 2.0符合性的關系
歐盟醫療器械指令93/42/EEC之條款4(5)明確規定:如果醫療器械也需要符合其他關于別的因素的指令,且也要求加貼CE標志時,(產品上)加貼的CE標志應表明該醫療器械也滿足這個指令的條款要求。
歐盟體外診斷醫療器械指令 98/79/EC之條款4(5)明確規定:如果醫療器械也需要符合其他關于別的因素的指令,且也要求加貼CE標志時,(產品上)加貼的CE標志應表明該醫療器械也滿足這個指令的條款要求。
即有源醫療器械上加貼了CE標志,表明制造商在聲明該醫療器械產品既符合對應的醫療器械指令的要求,也符合其他適用的CE標志指令的要求,如RoHS2.0的要求。
表述的更容易理解一點就是:
不符合RoHS指令的有源醫療器械產品,不能加貼CE標志;
加貼了CE標志但不符合RoHS的有源醫療器械產品,進入歐盟的話,是違法的,存在極大的風險;
不符合RoHS指令的有源醫療器械產品,不應該通過醫療器械CE認證。也即意味著對醫療器械的CE認證,應該審核RoHS符合性,并確定產品符合RoHS要求才允許通過認證。
對于醫療器械認證機構而言,讓不符合RoHS2.0要求的有源醫療器械產品通過CE認證將具有極大風險。如果經其認證的有源醫療器械在歐盟市場上被監管部門抽查,且結果是不符合RoHS2.0的要求,那么,認證機構的資質可能因此而受影響,因為產品的CE標志上帶有認證機構的編號。
因此,歐盟有源醫療器械的認證機構在開展CE認證時,應該審核產品的RoHS2.0符合性。
三、醫療器械CE認證審核員如何把握產品對RoHS2.0的符合性
1.準確把握與CE標志相關的RoHS2.0要求
a)產品中有害物質不能超標
b)產品需制訂并隨附技術文檔,技術文檔需符合EN 50581標準的要求
c)產品隨附符合性聲明,聲明需符合RoHS2.0附件VI的格式和內容要求
d)產品加貼CE標志,標志格式需符合歐盟對CE標志的要求
2. 重點關注產品有害物質超標的主要風險
a) 風險材料:是導致產品有害物質超標的最大風險。但有害物質只會存在于某些材料中,見下表:
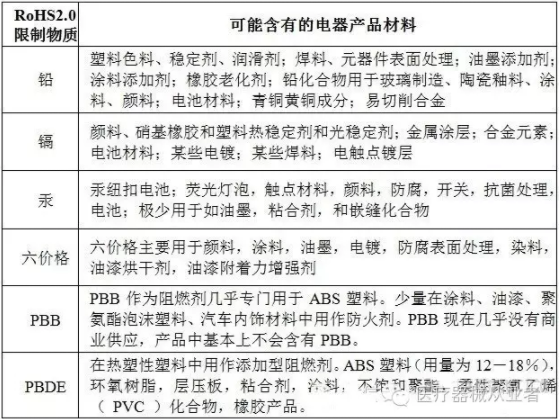
c)人為失誤:如混料/混貨:回用塑料混料;非常多,風險最大,同時做兩種產品的要重點關注
3. 把握關鍵風險過程
如上所述,有害物質超標的風險,最主要的來源是原材料中有害物質超標。所以,一下過程的控制效果是關鍵,審核員應重點關注這些過程:
物料確認過程:必須確認公司擬采購的物料是經過充分驗證符合RoHS的。
采購過程:公司采購的產品只能是經過確認的產品,供應商也必須是經過確認的。
來料檢驗:公司應該具備一定的有害物質檢測能力,如配備XRF類快速測定儀。
倉庫管理:確保物料的正確標識、正確發放,確保不發生混料。
生產過程:確保物料的正確領用,確保不發生混料。
產品檢測:一般客戶要求產品進行年度第三方RoHS檢測。
技術文檔:確保公司已按照EN 50581要求制定了技術文檔。
符合性聲明:確保公司已按照RoHS2.0附件VI的格式和內容制定了符合性聲明。
CE標志:確保CE標志格式和加貼的正確性。

